
Abstract
Suppressing infections of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) will probably require the rapid identification and isolation of individuals infected with the virus on an ongoing basis. Reverse-transcription polymerase chain reaction (RT–PCR) tests are accurate but costly, which makes the regular testing of every individual expensive. These costs are a challenge for all countries around the world, but particularly for low-to-middle-income countries. Cost reductions can be achieved by pooling (or combining) subsamples and testing them in groups1,2,3,4,5,6,7. A balance must be struck between increasing the group size and retaining test sensitivity, as sample dilution increases the likelihood of false-negative test results for individuals with a low viral load in the sampled region at the time of the test8. Similarly, minimizing the number of tests to reduce costs must be balanced against minimizing the time that testing takes, to reduce the spread of the infection. Here we propose an algorithm for pooling subsamples based on the geometry of a hypercube that, at low prevalence, accurately identifies individuals infected with SARS-CoV-2 in a small number of tests and few rounds of testing. We discuss the optimal group size and explain why, given the highly infectious nature of the disease, largely parallel searches are preferred. We report proof-of-concept experiments in which a positive subsample was detected even when diluted 100-fold with negative subsamples (compared with 30–48-fold dilutions described in previous studies9,10,11). We quantify the loss of sensitivity due to dilution and discuss how it may be mitigated by the frequent re-testing of groups, for example. With the use of these methods, the cost of mass testing could be reduced by a large factor. At low prevalence, the costs decrease in rough proportion to the prevalence. Field trials of our approach are under way in Rwanda and South Africa. The use of group testing on a massive scale to monitor infection rates closely and continually in a population, along with the rapid and effective isolation of people with SARS-CoV-2 infections, provides a promising pathway towards the long-term control of coronavirus disease 2019 (COVID-19).
Similar content being viewed by others

Main
COVID-19 represents a major threat to global health. Rapidly identifying and isolating individuals with SARS-CoV-2 infections is one of the most important available strategies for containing the virus. However, each diagnostic test12 for the SARS-CoV-2 virus costs US$50–100. Therefore, testing individuals regularly—which may be required to eliminate the virus—is expensive. The costs are unaffordable for most low-income countries, which have limited available resources for large-scale SARS-CoV-2 testing. It is therefore important to investigate whether there are more-efficient ways to identify those individuals infected with the virus.
The first step in testing—swab collection—is labour-intensive but does not require expensive chemicals or equipment. It may therefore be feasible to collect swabs regularly from everyone. The next step involves RT–PCR machines13. These require expensive chemical reagents, which are currently in short supply, as well as skilled personnel. To reduce the cost, we need to minimize the total number of tests. The speed of testing is also a key concern because SARS-CoV-2 is so infectious. Each RT–PCR test takes several hours in the laboratory, time during which the virus can spread14.
To identify individuals infected with SARS-CoV-2, the naive approach is to test everyone separately—that is, to perform one test per person. However, at low prevalence it is far more efficient to pool (or combine) samples and test these sample pools together. This idea of group testing was proposed by Dorfman in 19431. At low viral prevalence p, Dorfman’s algorithm reduces the number of tests per person to ≈ 2√p (Supplementary Information I). The algorithm that we present is more efficient, as it requires only ≈ epln(1/p) tests per person at low p, where e = 2.718… is Euler’s number (Supplementary Information II). As an example, a survey of private residential households in England and Wales, released on 4 September 2020 by the Office of National Statistics15,16,17, estimated a prevalence of p = 0.05% (95% confidence interval, 0.04–0.07%). For p = 0.05%, Dorfman’s algorithm offers a 22-fold cost reduction whereas ours offers a 100-fold cost reduction. The main obstacle to achieving these large cost savings is the number of samples that can be pooled without compromising detection. Here we present proof-of-concept experiments that show that one positive sample in a pool of a hundred samples can still be reliably detected. We also discuss how the pool size could be further increased to obtain the full benefits of group testing at low prevalence. If larger pool sizes and the associated cost reductions can be achieved, group testing may provide an affordable pathway to the long-term control of SARS-CoV-2.
In this paper, our focus is on population screening and not on protocols for use with at-risk groups or in clinical settings. The prevalence is typically much higher among individuals in at-risk groups or among individuals who present themselves for testing. For example, in the week ending 2 September 2020, 0.6% of the tests performed in hospitals in England were positive18, which suggests a prevalence that is an order of magnitude greater than that in the wider population, quoted above. In addition, in clinical settings the overriding concern should be to test the individual patient as quickly and accurately as possible. In most situations, that means performing an individual test. We are not suggesting the use of group testing as a strategy for testing patients in clinics, especially those with symptoms.
With this caveat, there are many potential applications of our method—for example, to screen sports teams whose players and staff must be tested regularly. A prominent rugby team in South Africa is now trialling our method. Early results indicate cost savings of more than an order of magnitude, with the successful detection of positive samples in groups of 81. Other applications include screening of staff and residents in care homes or pre-flight screening of passengers for commercial flights. The Government of Rwanda has adopted group testing as a national strategy and all air passengers are required to undergo a group test locally. This has helped to revive tourism in the country. Regular screening of university halls of residence, laboratories or departments could similarly enable safer in-person interactions. There is also potential for combining group testing with cheaper multiplex RT–PCR tests19 . The combination could reduce the costs of population-wide screening by more than two orders of magnitude compared with current methods.
Group-testing algorithms generally require more than one round of testing. In Dorfman’s algorithm, a first round of group tests is followed by a second round in which each member of every positive group is tested individually. Our algorithm involves a similar first round of group tests, although with a larger group size. Positive groups proceed to a second round of ‘slice tests’, which usually suffices to identify all individuals who are infected, without any need for individual tests. Occasionally one and, very rarely, more than one additional round of slice tests are required. We compare our approach with other approaches in detail in the Supplementary Information. There are adaptive algorithms that require fewer tests but more rounds of testing, during which time viral prevalence can grow. Such searches are disfavoured at low prevalence (see ‘Largely parallel searches are preferred’). There are also non-adaptive algorithms that require only one round of testing4,5,6,7,11. Although these algorithms appear attractive, they have disadvantages compared with our algorithm—for example, a higher failure rate (Supplementary Information sections III, IX). In our approach, the first round of tests—which are performed on groups—provides a valuable ‘sanity check’ on the viral prevalence in the population, before the second round of more-numerous slice tests is performed. More generally, group tests can provide a highly efficient means of tracking the viral prevalence in various populations in real time (Supplementary Information VIII).
Group testing is most obviously effective when none of the group members is infected: just one test suffices to clear everyone. Our algorithm takes full advantage of this powerful result. In the first round of tests, subsamples from all group members are pooled and tested together. For our algorithm, the optimal group size is N ≈ 0.35/p. The expected number of group members who are infected is 0.35 and a group will test negative more than 70% of the time. Groups that test positive are passed on to the first round of slice tests, which we describe next.
When one member of a group is infected
Consider the case in which only one member of the group is infected. The idea behind our algorithm is geometrical: the group of individuals to be tested is represented by a set of N points on a cubic lattice in D dimensions, organized in the form of a hypercube with L points on a side (Fig. 1), so that
Instead of directly testing the samples taken from every individual, we first divide each of them into D equal subsamples. These DN subsamples are recombined as follows. Slice the hypercube into L planar slices, perpendicular to one of the principal directions on the lattice. Form such a set of slices in each of the D principal directions and pool the LD − 1 = N/L subsamples that correspond to each slice. Altogether, DL slices are tested in parallel, in each round of slice tests. If there is one individual who is infected, then one slice out of the L slices in each of the D directions will yield a positive result. That slice indicates the coordinate of the individual in the corresponding principal direction.

Each vertex represents an individual. The hypercube is sliced into L slices, in each of the D principal directions. Samples from N/L individuals are pooled into a sample for each slice. For this example, the three sets of slices are shown in blue, red and green. If, among the N individuals being screened, only one is infected, tests on each set of slices identify their coordinate in that direction. Thus, in this example, only nine tests uniquely identify them. As the viral prevalence p decreases, the optimal group size N, the dimension D and the efficiency gain increase.
Therefore, the total number of tests required to find the individual who is infected is
where we used equation (1). Treating D as a continuous variable, the right side of equation (2) diverges at both small and large D, possessing a minimum at
corresponding to L = e and a total of eln(N) tests. In reality, D and L must be integers, but using L = 3 achieves almost the same efficiency (in the total number of tests, e is replaced with 3/ln(3) ≈ 2.73, which is less than 0.5% larger, whereas using L = 2 or 4 gives 2/ln(2) = 4/ln(4) ≈ 2.89, which is more than 5% larger). With no further constraint, finding one person who is infected in a population of a million—using L = 3—requires only 39 tests, performed in one round of testing. To understand this calculation, note that 313 > 106; therefore, a hypercube of side L = 3 with dimension D = 13 contains more than a million points. A round of slice tests on this hypercube consists of DL = 13 × 3 = 39 slice tests.
Proof of concept
In real-time PCR tests, the target RNA molecules are reverse-transcribed into DNA, which is replicated exponentially until it can be detected using fluorescence. In a perfectly efficient test, the number of DNA molecules doubles in every PCR cycle. The test is extremely sensitive: fewer than 10 molecules of viral RNA are sufficient for a positive test result13. A nasopharyngeal swab taken in the first 5 days of symptoms yields, on average, around 2 × 105 viral RNA molecules per millilitre20. Asymptomatic individuals who are infectious appear to have similar viral loads21. In the usual testing protocol, just 5 μl of the solution, containing on average around 1,000 RNA molecules, is included in the mix that is then analysed by PCR. Samples taken at earlier or later stages of infection, or in younger patients whose antibodies have suppressed the virus, have fewer virus particles present. In practice, this reduced number of virus particles is thought to be the most important cause of testing error, taking the form of false-negative test results17,22,23. In pooled testing, positive subsamples are diluted with negative subsamples. Dilution by 100-fold, for example, means that on average only around 10 RNA molecules are likely to be present in the RT–PCR test. In principle, this should still be sufficient to yield a positive result.
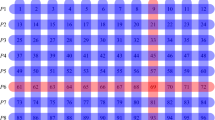
As a proof of concept, using oropharyngeal swab specimens collected during COVID-19 surveillance in Rwanda, we investigated whether known positive specimens still tested positive after they were diluted 20-, 50- or 100-fold through pooling with negative specimens (Methods). We used a RT–PCR test that targets the N and orf1ab genes of SARS-CoV-2, a combination that is used routinely for diagnostic screening for SARS-CoV-2 infections in Rwanda. The standard protocol is to consider a test positive if PCR amplification produces an above-background fluorescence signal for both target genes at a PCR cycle number—that is, a cycle-threshold (Ct) value—of Ct ≤ 40. Our key finding is that typical positive specimens can still be detected even after dilution by up to a 100-fold (Fig. 2). Previously published experiments9,10,11 have demonstrated detection after 30-, 32- and 48-fold dilution. As a consistency check, we determined the change in the Ct value (ΔCt) when going from a 50- to a 100-fold dilution. As noted above, a positive sample diluted 100-fold in principle requires one more cycle of PCR amplification than when diluted 50-fold to achieve the same fluorescence signal, implying ΔCt ≈ 1.0. Consistent with this expectation, we find ΔCt ≈ 1.0 ± 0.15 (mean ± s.d.) for the N gene and ΔCt ≈ 1.1 ± 0.14 for the orf1ab gene. The changes in Ct values for other dilutions are also consistent with this interpretation.

a, b, Each of six typical SARS-CoV-2-positive specimens was diluted through pooling with 19, 49 or 99 negative specimens. A Ct value (that is, the PCR cycle at which the fluorescence signal generated by a specimen exceeds the baseline signal) was determined for each pool through RT–PCR amplification of the N (a) and orf1ab (b) genes of SARS-CoV-2. For each gene, the Ct values are plotted against the dilution factor. The red horizontal lines indicate the Ct value (40) at or below which a specimen is considered positive. All Ct curves stay below the red lines even if the positive specimens are diluted 100-fold (Extended Data Fig. 1 and Extended Data Table 2).
We estimated the postdilution sensitivities by combining (technically, convolving) the probability distribution for predilution Ct values for positive samples (Extended Data Table 3) with the probability distribution for the increase in ΔCt as inferred above. Treating both distributions as Gaussian, the distribution of postdilution Ct values is also Gaussian, with the mean given by the sum of the means and the variance given by the sum of the variances. In this way, we estimated that a 40-cycle PCR test targeting the SARS-CoV-2 N (or orf1ab) gene, respectively, has postdilution sensitivities for 20-, 50- and 100-fold dilutions of 91%, 88% and 85% (or 85%, 81% and 77%, respectively). We have confirmed these estimates using two additional datasets. First, we used an independent sample of Ct values for 107 positive specimens collected in Rwanda using tests that targeted the same two genes. Second, we reanalysed a published dataset of 26 positive specimens from a recent study of pooled testing for SARS-CoV-2 in Israel10, in which a single gene (the E gene) was targeted. All three datasets gave broadly consistent results.
The positive samples that are most likely to be missed because of dilution are those with the highest Ct values before dilution—that is, those with the lowest viral load. The individuals concerned are likely to be the least infectious24,25. Conversely, those individuals—whether symptomatic or asymptomatic—whose samples have the lowest Ct values, which are the least affected by sample dilution, are the most important to detect as they are likely to be the most infectious. Nevertheless, it is important to consider ways in which the loss in sensitivity due to dilution might be mitigated. The most obvious is to re-test sufficiently often (say, every 3 days) to ensure a test occurs in the period of highest viral abundance, for any individual infected with SARS-CoV-2. Group tests involve the greatest degree of dilution in our method, but they are also the cheapest testing stage to repeat frequently and to thereby mitigate sensitivity loss. Likewise, one could increase the number of PCR cycles to 44, the maximum used in the previous study10. Similarly, the volume of the sample used in the RT–PCR test can be increased from 5 μl to 10 μl, reducing the Ct value by one (this is done in the laboratory that we are working with in South Africa). Furthermore, the viral concentration in the pooled sample might be increased by physical or chemical methods such as ultracentrifugation or precipitation. Finally, PCR machines might be re-engineered to enable larger sample volumes to be tested. All of these possibilities are worth exploring.
When more than one member of a group is infected
So far, we have assumed that only one member of the group is infected. We must also consider what happens when two, three or more members of a group are infected. We will have to discover that number in the course of the slice tests. A feature of group testing is that the first round of group tests, which are relatively few in number, allows us to conveniently update our knowledge of the viral prevalence p, before any individuals who are infected have been identified (Supplementary Information section VIII).
Given p, the probability that k members of a group of size N are infected is described by a Poisson distribution with mean λ = pN. For λ well below unity, the probability decreases rapidly with increasing k. At very low p, the optimal N is very large, so D = logL[N] ≫ 1. The advantages of the hypercube algorithm are particularly clear in this limit. Therefore, we describe this limit first before discussing realistic values of D. In this section we asssume, for simplicity, perfect accuracy of all tests.
The first round of slice tests—as described above—yields, for L = 3, a set of triples of zeros and ones, that is, {1, 0, 0}, {1, 1, 0} or {1, 1, 1} or a permutation thereof, for every principal direction of the lattice. Let σ be the sum of the three values (so σ = 1, 2 or 3) and dσ the number of directions in which the value σ occurs, so d1 + d2 + d3 = D. For D ≫ 1, the number of group members who are infected (k) may be accurately inferred from the observed values of dσ, even before any individuals who are infected are identified. Knowing k, we then find all individuals who are infected as follows. First, if k = 1, then d1 = D. Each positive slice indicates the coordinate value in that direction. Thus, the individual who is infected is identified in one round of slice tests. Second, suppose k = 2, then d2 > 0 but d3 = 0. If d2 = 1, the two individuals who are infected are immediately identified. If d2 > 1, choose one of the directions with σ = 2, and treat the two positive slices as smaller hypercubes, each containing one individual who is infected. A further round of slice tests identifies one and the other is found by elimination. Third, if k = 3 then, at large D, at least one direction has σ = 3. Choose one such direction and treat two of the positive slices as smaller hypercubes, each containing one individual who is infected. A slice test on each identifies two individuals who are infected and the third is found by elimination; if k > 3, the number of rounds of slice tests required to identify all individuals who are infected is slightly larger than k. However, for the optimal value of group size, the probability to have k > 3 members who are infected is negligibly small.
Thus, in the large D limit, to a good approximation k rounds of slice tests suffice to identify k individuals who are infected. In the Supplementary Information, we show that, at low prevalence p, assuming Poisson statistics, the expected number of tests per person ⟨T⟩/N that is required to identify all individuals who are infected is minimized for N ≈ 0.350/p. At this optimal group size, ⟨T⟩/N ≈ epln(0.734/p) (Fig. 3). The reciprocal of this number is the efficiency gain—that is, the cost savings factor—relative to testing every individual.

The results are shown on a log–log plot. The dashed grey line shows epln(0.734/p), the result obtained in the large D approximation, for which the optimal group size N ≈ 0.35/p. The coloured lines show the results obtained from a detailed analysis when the group size N = 3D with D an integer. Where 0.35/p is an exact power of 3, as at the left end of each coloured curve, optimal performance is attained. As p is increased, a growing fraction of sites in the 3D hypercube are left empty, until 0.35/p is again an exact power of 3 (Supplementary Information).
For practical applications, we are interested in the efficiency of the algorithm at modest values of D such as 3, 4 or 5. This requires a more intricate analysis, the details of which we provide in the Supplementary Information. However, some simple and general statements are included here. First, when all directions yield σ = 1, only one individual is infected and they are immediately and uniquely identified. This is the most probable outcome of the first round of slice tests. Second, when σ > 1 in only one direction then two (or three) individuals who are infected are uniquely identified without further tests. If σ > 1 in more than one direction, a second round of slice tests is needed. We can eliminate any slice that tested negative in the first round of slice tests and thus work with a smaller hypercube. We make only one approximation in our analysis, namely we assume the infected samples are rare in the hypercube. They may then be treated as independent, randomly chosen points. Within this approximation, we compute the probabilities through to the second round of slice tests. Notably, we find that the hypercube algorithm remains highly efficient at modest values of D. For example, for λ = 0.35 and D = 4, in 93.3% of cases one round of slice tests suffices to identify all individuals who are infected. For the remaining 6.7% of cases, one more round suffices in all but 0.01% of cases, a very low theoretical failure rate (which, we emphasize, does not include experimental errors). The expected total number of tests per person, for D = 3, 4 and 5, is plotted in Fig. 3. When 0.35/p is an exact power of 3, as is the case at the left end of each coloured curve in Fig. 3, the performance is best relative to the large D formula given in the previous paragraph. As p is increased, an increasing fraction of sites in the 3D hypercube are left empty until the next exact power of 3 is reached. Nevertheless, at the values of p shown, pooling always results in a high efficiency gain. As Fig. 3 shows, the large D approximation provides a surprisingly good (and very convenient) fit to the low D results (further details are provided in Supplementary Information sections IV–VII).
Largely parallel searches are preferred
Some search methods require fewer tests but more rounds of testing. A binary search2,3, for example, finds one individual among N in ~log2[N] tests, a factor of eln[2] ≈ 1.88 fewer tests than needed by our hypercube algorithm at large D. However, the tests must be performed serially, requiring ~log2(1/p) rounds of testing. For p = 0.4% (or 0.15%) a binary search takes 8 (or more than 9) rounds of testing whereas a hypercube search takes typically 2 and occasionally 3 rounds (in both cases). For a highly infectious disease such as COVID-19, saving time is crucial because individuals who are infected and still at large can infect others. The doubling time for COVID-19 has been estimated at τ2 ≈ 2 days14,26. If each testing round takes τ days, the viral prevalence in the population at large grows by \( \sim {(1/p)}^{\tau /{\tau }_{2}}\) during a binary search. If this growth factor exceeds eln(2), a binary search will do worse than a hypercube search. Assuming τ ≈ 1/3 day, we find that for p < 1%, the hypercube search is preferred. Another advantage of the hypercube search is that it includes many consistency checks. For example, finding σ = 1 in one direction and σ = 0 in the others indicates a testing error. By contrast, a binary search relies on repeated testing of the positive sample, so that a single false-negative result can prematurely terminate the search.
Conclusions
The hypercube algorithm offers an attractive compromise between minimizing the total number of tests to reduce costs and maximizing the speed of testing to reduce the spread of the virus. We have demonstrated its viability for group sizes up to 100 samples, showing that cost savings of a factor of nearly 20 can, in principle, already be achieved. We have quantified the loss of sensitivity due to dilution and discussed a number of ways in which it may be mitigated—for example, through frequently repeated group tests. These strategies could enable the use of larger pool sizes, bringing even greater cost savings at low prevalence. The most striking aspect of our approach is how rapidly the cost of testing a population can fall, pooled test sensitivity permitting, as the viral prevalence decreases. This should incentivise decision-makers to act firmly to drive the prevalence down through mass screening, contact tracing and isolation. As the viral prevalence is reduced, all aspects of this strategy become more and more affordable.
Methods
Observational study design
We conducted an experiment to evaluate the hypothesis that known SARS-CoV-2-positive oropharyngeal swab specimens collected during COVID-19 surveillance in Rwanda will test positive after they are combined with as many as 99 known SARS-CoV-2-negative specimens. This was followed by an observational study that aimed to apply our hypercube algorithm to increase the efficiency of community testing for COVID-19 in Rwanda. In the experiment, two different sets of sample pools were tested for SARS-CoV-2 using RT–PCR. Each set consisted of three sample pools that contained one known SARS-CoV-2-positive sample diluted in ratios of 1:20, 1:50 and 1:100 by combining it with equivalent amounts of 19, 49 and 99 known SARS-CoV-2-negative samples, respectively (Fig. 2 and Extended Data Table 2). In the observational study, 1,280 individuals selected from the community were tested for SARS-CoV-2 using RT–PCR. One third of the individuals were participants in a screening for severe acute respiratory infections and influenza-like illness conducted in 30% of the health facilities found across the 30 districts of Rwanda. The remaining two thirds were from COVID-19 screening of at-risk groups in the capital city of Kigali. The latter group consisted mainly of individuals (market vendors, bank agents and supermarket agents) who remained active during the lockdown imposed by the Government of Rwanda to contain COVID-19. Extended Data Table 1 summarizes the characteristics of the study participants.
The positive fraction of RT–PCR tests for SARS-CoV-2 conducted in Rwanda in March 2020 suggests an upper bound of 2% for the virus prevalence in the country. Using p = 2% in the hypercube algorithm indicated an optimal sample group size of 17.5. For convenience, the 1,280 individual samples were combined in 64 groups of 20 samples before testing for SARS-CoV-2 (Extended Data Fig. 1).
We used two established experimental protocols for SARS-CoV-2 testing. The first is a protocol by DAAN Gene and Sun Yat-sen University that is available online (https://prolabcorp.com/daan-rt-pcr-reagent-set-for-covid-19-real-time-detection-for-48-samples-research-use-only), and is also under review by the WHO. The second protocol13 is widely used by the scientific community. The first protocol is used for routine screening for SARS-CoV-2, whereas the second protocol is used only if the first protocol produces a positive result and confirmation is therefore required.
Sample collection and pool design
Oropharyngeal swabs were collected by wiping the tonsils and posterior pharynx wall with two swabs, and the swab heads were immersed in 3 ml viral transport medium. Samples were transported in viral transport medium to the Rwanda National Reference Laboratory immediately after collection. Samples that had to be transported over a long distance were stored in dry ice. Each sample had a volume of 3 ml, of which an aliquot of 200 μl was used for pooled testing, and the remainder was temporarily stored at −20 °C until the result of the pooled testing was known. The aliquot (200 μl) of each sample was mixed with aliquots with the same volume of other samples of the same pool in a Falcon 15-ml conical tube and, after vortexing for 5 s, 200 μl of the mixture was pipetted for downstream RNA extraction. Then, 5 μl of the extracted RNA was added to 20 μl of master mix for a total of 25 μl to be amplified by RT–PCR. If a pool tested positive, stored samples from that pool were processed to identify the positive samples. Individual samples were barcoded, making it easy to trace individuals that tested positive and minimizing the risk of confusion of samples. Pool design and subsequent experimental analysis (see ‘RT–PCR for SARS-CoV-2’) were implemented with the aid of a robot to reduce human error.
RT–PCR for SARS-CoV-2
Total viral RNA was extracted from swab specimens using the QIAamp Viral RNA 91 Mini Kit (QIAGEN), according to the manufacturer’s instructions. RNA samples were screened for SARS-CoV-2 using a 2019-nCoV RNA RT–PCR test that targets two genes that, respectively, encode an open reading frame (denoted orf1ab) and a nucleocapsid protein (denoted N) (DAAN Gene and Sun Yat-sen University). For orf1ab, CCCTGTGGGTTTTACACTTAA and ACGATTGTGCATCAGCTGA were used as forward and reverse primers, respectively, together with a 5′-VIC-CCGTCTGCGGTATGTGGAAAGGTTATGG-BHQ1-3′ probe. For N, GGGGAACTTCTCCTGCTAGAAT and CAGACATTTTGCTCTCAAGCTG were used as forward and reverse primers, respectively, together with a 5′-FAM-TTGCTGCTGCTTGACAGATT-TAMRA-3′ probe. The RT–PCR reaction was set up according to the manufacturer’s protocol, with a total volume of 25 μl. The reaction was run on the ABI Prism 7500 SDS Instrument (Applied Biosystems) at 50 °C for 15 min for reverse transcription, denatured at 95 °C for 15 min, followed by 45 PCR cycles of 94 °C for 15 s and 55 °C for 45 s. A threshold cycle (Ct) ≤ 40 indicated a positive test; Ct > 40 indicated a negative test. Positive controls for the reaction showed amplification as determined by curves for FAM- and VIC-detection channels, and Ct ≤ 32. Positive tests were confirmed using LightMix SarbecoV E-gene and LightMix Modular SARS-CoV-2 RdRp RT–PCR tests that target the envelope (E) and RNA-directed RNA polymerase (rdrp) genes, respectively, as described by the manufacturer (TIB MOLBIOL). Both the primers used and the RT–PCR reaction conditions were previously described13.
Statistical analysis
Ct values were tested for normality using the Shapiro–Wilk test. A confidence bound for a sample of n Ct values was calculated as \({\bar{C}}_{{\rm{t}}}\pm {t}_{{\rm{df}}}^{\ast }\times s\), where \({\bar{C}}_{{\rm{t}}}\) is the sample mean, s is the sample standard error of the mean and \({t}_{{\rm{d}}{\rm{.f}}.}^{\ast }\) is an appropriate quantile of the Student’s t-distribution with n − 1 degrees of freedom (d.f.). A confidence bound for the sum of the means of two samples of Ct values of sizes n1 and n2, respectively, was calculated using the same formula, with \({\bar{C}}_{{\rm{t}}}\) set to the sum of the individual sample means, s set to the sum of the standard errors of the individual sample means and d.f. set to the smaller of n1 − 1 and n2 − 1. Statistical analysis was done using the R statistical computing environment (https://www.r-project.org/).
Loss of sensitivity due to dilution
To estimate the postdilution sensitivities of RT–PCR tests with different maximum numbers of PCR cycles, we combined two datasets. First, we used the mean and standard deviations of the number of additional PCR cycles required for a positive detection, after a k-fold dilution of a positive specimen (Fig. 2, showing the data in Extended Data Table 3). Second, we used the mean and standard deviation of Ct values for positive specimens sampled from a target population. We combined (or, more accurately, convolved) the two probability distributions, represented as Gaussians to calculate the sensitivity of a ≤x cycle PCR test as the probability that the Ct value of a k-fold diluted positive specimen sampled from the same population will be ≤x. Using a representative sample of 33 positive specimens identified during clinical screening for SARS-CoV-2 in Rwanda (Extended Data Table 3), we estimate that a ≤40-cycle PCR test targeting the SARS-CoV-2 N (or orf1ab) gene, respectively, has postdilution sensitivities for 20-, 50- and 100-fold dilutions of 95%, 92% and 89% (or 86%, 82% and 77%, respectively). For a ≤44-cycle PCR test targeting the N (or orf1ab) gene, we obtain postdilution sensitivities for 20-, 50- and 100-fold dilutions of 99%, 98% and 98% (or 96%, 94 and 92%), respectively. As mentioned in the paper, a maximum of 44 PCR cycles was used in the recent study of pooled testing for SARS-CoV-2 in Israel10.
As further checks, we applied the same analysis to (1) an independent sample of 107 positive specimens collected in Rwanda and (2) the previously published dataset10, which consists of the 26 positive specimens identified in the previous study10. From the Rwandan dataset, we estimated that a 40-cycle PCR test targeting the N (or orf1ab) gene, respectively, has postdilution sensitivities for 20-, 50- and 100-fold dilutions of 91%, 88% and 85% (or 85%, 81 and 77%, respectively). For a 44-cycle PCR test targeting the N (or orf1ab) gene, respectively, the predicted sensitivities are 97%, 96% and 95% (or 94%, 92 and 90%). The previously published dataset10 contains Ct values for only one gene—the E gene of SARS-CoV-2. On the basis of the arguments described above, we assume for simplicity that diluting a positive specimen by 20-, 50- and 100-fold adds approximately 5, 6 and 7, respectively, to the original Ct value. Applying these assumptions to the previously published dataset10, we infer postdilution sensitivities for 20-, 50- and 100-fold dilutions of 94%, 92% and 89%, respectively, for a ≤40-cycle PCR test, and 99%, 98% and 97%, for a ≤44-cycle PCR test. These results are comparable to those reported from our experiments. Together, these findings confirm that diluting positive samples does result in a loss of sensitivity, but that much of the loss can be offset by increasing the number of PCR cycles. In particular, sensitivities above 90% can be achieved for 100-fold dilution by using 44 PCR cycles, only 10% more than the number routinely employed.
Ethics approval
Ethics approval was obtained from the Rwanda National Ethics Committee (FWA Assurance No. 00001973 IRB 00001497 of IORG0001100/20March2020) and written informed consent was obtained from the participants.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
All data that support the findings of this study are available from the corresponding authors upon reasonable request.
Code availability
All codes that were used in the study are available from the corresponding authors upon reasonable request.
Change history
16 June 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41586-021-03630-z
References
Dorfman, R. The detection of defective members of large populations. Ann. Math. Stat. 14, 436–440 (1943).
Hwang, F. K. A method for detecting all defective members in a population by group testing. J. Am. Stat. Assoc. 67, 605–608 (1972).
Allemann, A. in Information Theory, Combinatorics, and Search Theory. Lecture Notes in Computer Science Vol. 7777 (Aydinian, H. et al.) 569–596 (Springer, 2013).
Aldridge, M., Johnson, O. & Scarlett, J. Group testing: an information theory perspective. Found. Trends Commun. Inf. Theory 15, 196–392 (2019).
Ghosh, S. et al. A compressed sensing approach to group-testing for COVID-19 detection. Preprint at https://arxiv.org/abs/2005.07895 (2020).
Heng, B. W. & Scarlett, J. Non-adaptive group testing in the linear regime: strong converse and approximate recovery. Preprint at https://arxiv.org/abs/2006.01325 (2020).
Broder, A. Z. & Kumar, R. A note on double pooling tests. Preprint at https://arxiv.org/abs/2004.01684 (2020).
Arevalo-Rodriguez, I. et al. False negative results of initial RT-PCR assays for COVID-19: a systematic review. Preprint at https://doi.org/10.1101/2020.04.16.20066787 (2020).
Lohse, S. et al. Pooling of samples for testing for SARS-CoV-2 in asymptomatic people. Lancet Infect. Dis. 20, 1231–1232 (2020).
Yelin, I. et al. Evaluation of COVID-19 RT-qPCR test in multi sample pools. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa531 (2020).
Shental, N. et al. Efficient high-throughput SARS-CoV-2 testing to detect asymptomatic carriers. Sci. Adv. 6, eabc5961 (2020).
Medicare. Medicare Administrative Contractor (MAC) COVID-19 Test Pricing. https://www.cms.gov/files/document/mac-covid-19-test-pricing.pdf (2020).
Corman, V. M. et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 25, 2000045 (2020).
Sanche, S. et al. High contagiousness and rapid spread of severe acute respiratory syndrome coronavirus 2. Emerg. Infect. Dis. 26, 1470–1477 (2020).
Office of National Statistics. Coronavirus (COVID-19) Infection Survey pilot: England and Wales, 4 September 2020. https://www.ons.gov.uk/peoplepopulationandcommunity/healthandsocialcare/conditionsanddiseases/bulletins/coronaviruscovid19infectionsurveypilot/englandandwales4september2020 (UK Department of Health & Social Care, 2020).
Pouwels, K. B. et al. Community prevalence of SARS-CoV-2 in England: results from the ONS Coronavirus Infection Survey Pilot. Preprint at https://doi.org/10.1101/2020.07.06.20147348 (2020).
Office of National Statistics. COVID-19 Infection Survey (Pilot): Methods and Further Information: Test Sensitivity and Specificity. https://www.ons.gov.uk/peoplepopulationandcommunity/healthandsocialcare/conditionsanddiseases/methodologies/covid19infectionsurveypilotmethodsandfurtherinformation#test-sensitivity-and-specificity (UK Department of Health & Social Care, 2020).
Office of National Statistics. Weekly Statistics for NHS Test and Trace (England) and coronavirus testing (UK): 27 August to 2 September. Annex Table 1: People newly tested for COVID-19 under Pillars 1 and 2, England. https://www.gov.uk/government/publications/nhs-test-and-trace-england-and-coronavirus-testing-uk-statistics-27-august-to-2-september-2020/weekly-statistics-for-nhs-test-and-trace-england-and-coronavirus-testing-uk-27-august-to-2-september#annex-table-1 (UK Department of Health & Social Care, 2020).
Reijns M. A. M. et al. A sensitive and affordable multiplex RT-qPCR assay for SARS-CoV-2 detection. Preprint at https://doi.org/10.1101/2020.07.14.20154005 (2020).
Wölfel, R. et al. Virological assessment of hospitalized patients with COVID-2019. Nature 581, 465–469 (2020).
Long, Q. X. et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat. Med. 26, 1200–1204 (2020).
Watson, J., Whiting, P. F. & Brush, J. E. Interpreting a covid-19 test result. Br. Med. J. 369, m1808 (2020).
Kucirka, L. M., Lauer, S. A., Laeyendecker, O., Boon, D. & Lessler, J. Variation in false-negative rate of reverse transcriptase polymerase chain reaction-based SARS-CoV-2 by time since exposure. Ann. Intern. Med. 173, 262–267 (2020)
Jaafar, R. et al. Correlation between 3790 qPCR positives samples and positive cell cultures including 1941 SARS-CoV-2 isolates. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa1491 (2020).
Service, R. F. One number could help reveal how infectious a COVID-19 patient is. Should test results include it? Science https://www.sciencemag.org/news/2020/09/one-number-could-help-reveal-how-infectious-covid-19-patient-should-test-results (2020).
Afshordi, N., Holder, B., Bahrami, M. & Lichtblau, D. Diverse local epidemics reveal the distinct effects of population density, demographics, climate, depletion of susceptibles, and intervention in the first wave of covid-19 in the United States. Preprint at https://arxiv.org/abs/2007.00159 (2020).
Acknowledgements
We thank the Rwanda Ministry of Health through RBC for discussions and correspondence, K. Smith and C. Squire for providing encouragement and references, and A. Jackson for discussions. Research at AIMS is supported in part by the Carnegie Corporation of New York and by the Government of Canada through the International Development Research Centre and Global Affairs Canada. Research at the Perimeter Institute is supported in part by the Government of Canada through the Department of Innovation, Science and Economic Development Canada and by the Province of Ontario through the Ministry of Colleges and Universities. The data collection, SARS-CoV-2 molecular experiments and analysis of the study were supported by the Government of Rwanda (Rwanda Biomedical Centre/Ministry of Health) and the Académie de Recherche et d’Enseignement Supérieur in collaboration with the University of Rwanda (ARES-UR Programme). All statistical and mathematical analyses were supported by the African Institute for Mathematical Sciences (AIMS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
L.M. and S.N. coordinated the experiments; P.N., T.N., E.N., M.S., C.M., D.N. and M.N. contributed to patient recruitment and data collection from the community; P.N., Y.B., J.S., A.U., R.R., E.L.N., E.M., N.R., I.E.M., J.B.M., C.M.M. and E.U. contributed to laboratory RT–PCR test validation, data analysis and interpretation. W.N. and N.T. contributed to the theory.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Maimuna Majumder, Sigrun Smola and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Amplification plot for sample pools.
Each of the 64 sample pools described in the text tests negative for SARS-CoV-2: the RT–PCR fluorescence curves show below-threshold net fluorescence values. By contrast, for both target genes of the positive control, the fluorescence curves cross the threshold after 32 PCR cycles. ΔRn denotes the difference between the fluorescence signal generated by a sample and a baseline signal. The yellow curves reaching ΔRn ≈ 2,000,000 and 1,750,000 represent the positive control for the N and orf1ab genes, respectively. The other yellow and orange curves represent internal controls.
Supplementary information
Supplementary Information
This file contains Supplementary Sections 1-9, including Supplementary Text and Data, Supplementary Figs 1-2 and Supplementary References.
Rights and permissions
About this article

Cite this article
Mutesa, L., Ndishimye, P., Butera, Y. et al. A pooled testing strategy for identifying SARS-CoV-2 at low prevalence. Nature 589, 276–280 (2021). https://doi.org/10.1038/s41586-020-2885-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2885-5
This article is cited by
-
High prevalence group testing in epidemiology with geometrically inspired algorithms
Scientific Reports (2023)
-
Adaptive group testing strategy for infectious diseases using social contact graph partitions
Scientific Reports (2023)
-
Evaluation of pooling strategy of SARS-CoV-2 RT-PCR in limited resources setting in Egypt at low prevalence
Comparative Clinical Pathology (2023)
-
Ferrobotic swarms enable accessible and adaptable automated viral testing
Nature (2022)
-
Identification-detection group testing protocols for COVID-19 at high prevalence
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
